
Understanding the enormous diversity of bacteriophages: The tailed phages that infect the bacterial family Enterobacteriaceae
Read the full article on ScienceDirect.
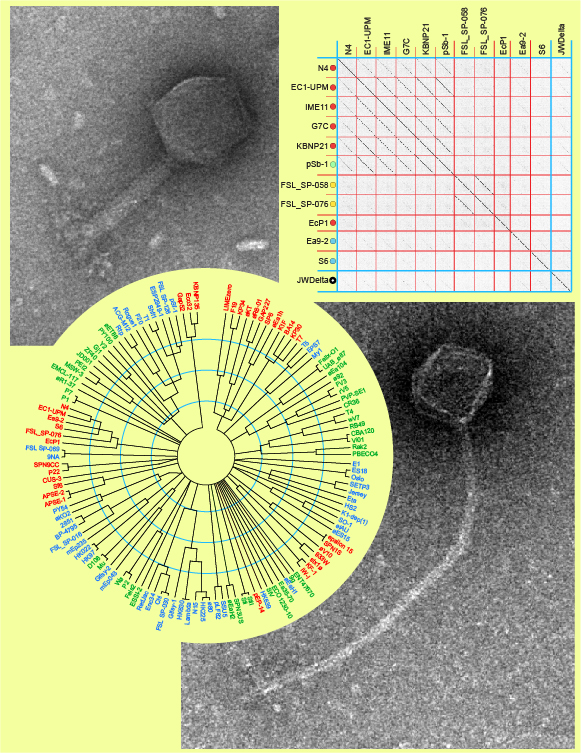
We have long-term interests in the diversity of E. coli/Salmonella phages (Casjens) and Erwinia phages (Grose) and wanted to know how the phages we have studied fit into the bigger picture of phage diversity. This caused us to realize that the big picture was not very clear at all and was seen by many as chaotic, with diversity that seemed beyond current understanding. We thus set out to see if we could make sense of the relationships among the known tailed phages that infect the Enterbacteriaceae bacterial family – the family that includes hosts for our phages of interest. The Enterbacteriaceae family consists of many well-known animal and plant pathogens and many well-studied phages infect these diverse hosts. Limiting the analysis to this single host family gave us enough diversity to examine, but it was not so large as to be completely overwhelming. We found that we could robustly place these phage genomes into 56 very different clusters and understand the few ‘outlier’ phages as hybrids between these clusters.
The major technical problems we had to overcome were the massive inconsistencies in the annotations of phage genomes. (1) Just finding all the 337 relevant tailed phage genomes was not trivial since such GenBank entries are not as standardized as one would like – they do not give the family of the host bacterium and some don’t give the host species(!). (2) Phage genome sequences are very often circular and authors open them more or less randomly for reporting a linearized version. (3) When comparing proteomes, a necessity in the analysis of more distant relationships, inconsistencies in annotation can drastically affect analyses. Thus a lot of manual labor went into generating a uniform set of genomes whose genes and proteins could be sensibly aligned and compared.
The major intellectual problems were rooted in the very essence of phage diversity and evolution. (1) Deciding on boundaries/definitions for relatedness was difficult because there is often a phage close to a boundary that questions its existence, and yet without that boundary, chaos ensues. (2) The immense amount of horizontal gene transfer can even make phages appear related that have quite different lifestyles. (3) The complexity of host range, including lack of host range studies as well as the ability of phages to switch hosts. Perhaps the biggest surprise was that horizontal exchange among tailed phages is not rapid enough to completely blur the lines between clusters. Further phage isolation will, undoubtedly, aid in our understanding of such issues.
We had something useful when we had compared enough genomes that a ‘new’ genome was often similar to one of the previously analyzed ones – i.e., there are in fact a limited number of phage genome ‘clusters’ (most phage are not hugely different from every other phage) and that we had enough genomes to identify a lot – perhaps the majority – of such clusters.
Introducing the authors


Julianne H. Grose, Sherwood R. Casjens
About the research
Understanding the enormous diversity of bacteriophages: The tailed phages that infect the bacterial family Enterobacteriaceae
Virology, Volumes 468–470, November 2014, Pages 421–443
Julianne H. Grose, Sherwood R. Casjens